



The full paper is available here.
On the Scalability of GNNs for Molecular Graphs
Scaling deep learning models has been at the heart of recent revolutions in language modeling and image generation. Practitioners have observed a strong relationship between model size, dataset size, and performance. However, structure-based architectures such as Graph Neural Networks (GNNs) are yet to show the benefits of scale mainly due to the lower efficiency of sparse operations, large data requirements, and lack of clarity about the effectiveness of various architectures.
We address this drawback of GNNs by studying their scaling behavior. Specifically, we analyze message-passing networks, graph Transformers, and hybrid architectures on the largest public collection of 2D molecular graphs. For the first time, we observe that GNNs benefit tremendously from the increasing width, number of molecules, number of labels, and diversity in the pretraining datasets.
We're excited to introduce MolGPS, a 1B parameter model for various molecular property prediction tasks. We hope that this work will pave the way for an era where foundational GNNs drive pharmaceutical drug discovery.
Model Details and Performance
MolGPS was trained on the LargeMix dataset mixture consisting of 5 million molecules grouped into 5 different tasks with each task having multiple labels. LargeMix contains datasets like the L1000_VCAP and L1000_MCF7 (transcriptomics), PCBA_1328 (bioassays), PCQM4M_G25 and PCGM4M_N4 (DFT simulations).
MolGPS was first pretrained using a common multi-task supervised learning strategy and was then finetuned (or probed) for various molecular property prediction tasks to evaluate performance. We benchmarked the performance of MolGPS on the Therapeutics Data Commons (TDC), MoleculeNet, and Polaris benchmarks.
Therapeutics Data Common (TDC)
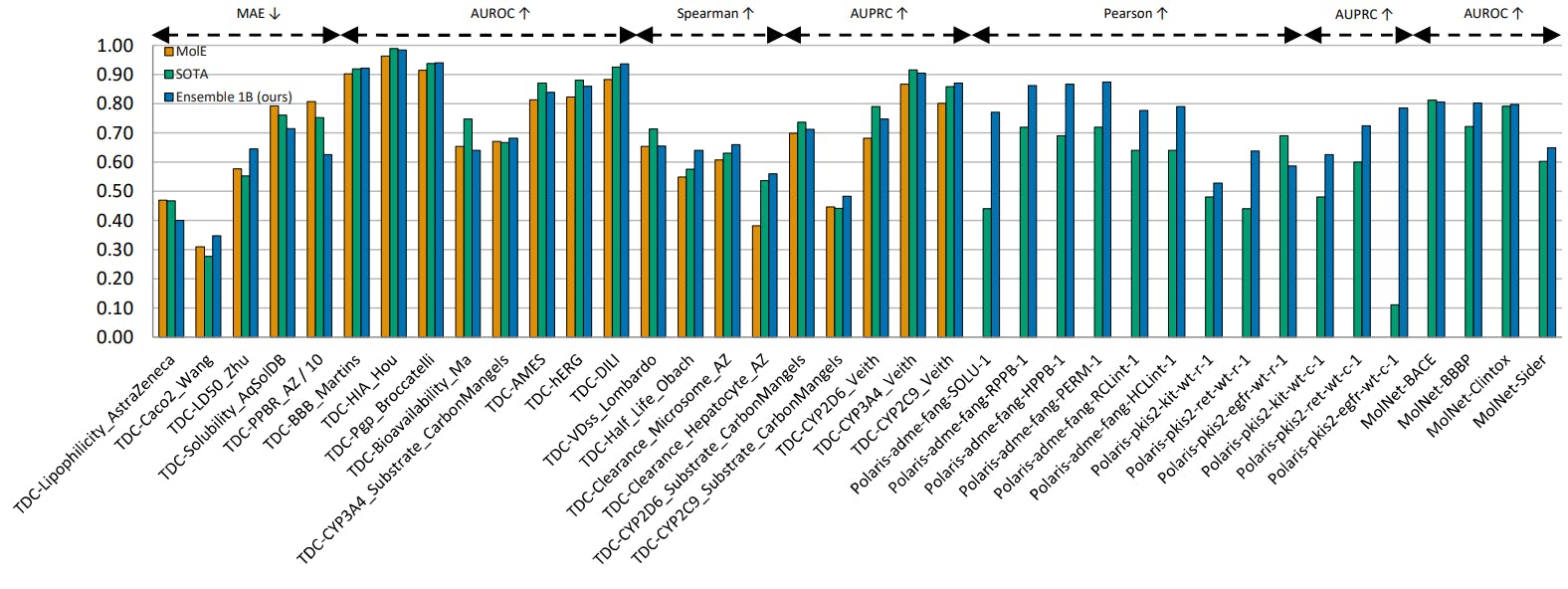
Our study focuses on the 22 ADMET (absorption, distribution, metabolism, excretion, and toxicity) tasks available in TDC. This benchmark has been around for years with continuous submissions from various groups, including both deep-learning models and traditional machine learning at the top of the benchmark, with a total of 8 models sharing the top positions across all 22 tasks. Simply by scaling our model, we found that MolGPS outperforms SOTA on 12/22 tasks.
We investigate 4 datasets from MoleculeNet that are commonly used in similar studies: BACE (that assesses the binding outcomes of a group of inhibitors targeting β-secretase), BBBP (the Blood-Brain Barrier Penetration that assesses if a molecule can penetrate the central nervous system), Clintox (which is relevant to the toxicity of molecular compounds, and Sider (the Side Effect Resource which contains information about adverse drug reactions in a database of marketed drugs). We found that MolGPS outperforms SOTA (all self-supervised or quantum-based self-supervised pre-trained models) on all 4 tasks.
Polaris
While TDC and MoleculeNet are commonly used benchmarks for open-source drug discovery evaluation, we note that they suffer from data collection and processing biases across dissimilar molecules. These limitations have been described previously in conversations throughout the community.
Polaris is a new collection of benchmarks and datasets curated through a standardized evaluation protocol developed by an industry consortium of biotechs and pharmaceutical companies. We investigated the performance of MolGPS on 12 ADMET and binding prediction tasks and found that MolGPS outperforms SOTA on 11/12 tasks.
Scaling Experiments
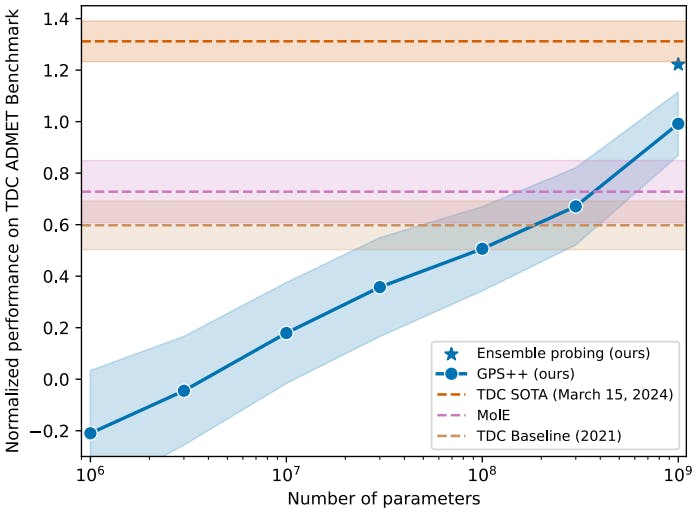
In the following experiments, we look at the performance of MolGPS while increasing the width such that the parameter count goes up from 1M to 1B parameters. To properly assess the benefits of scale, we evaluate the performance of the model when probing the 22 downstream tasks from TDC. Here, the hyper-parameter search is done with 10M parameters, and width scaling is done in a zero-shot setting using the muTransfer technique.
We also observe that GNNs benefit tremendously from increasing the model's width and that the gains are consistent and linear to the logarithm of the parameter count. Indeed, there appears to be no slowdown in the scaling curve, hinting that we could continue to improve the performance with larger and larger models, similar to those found in large language models (LLMs).
Further, we note that MolGPS shows significant improvement over the performance achieved by the TDC baselines. This represents the best model performance per task from when TDC was first introduced in 2021. Note that on the y-axis, a value of 0 represents the average of all submissions to the TDC benchmark. Compared to the latest SOTA on TDC, our model performs very competitively, and it is easy to imagine that we could pass the last line simply by scaling the model further. Below, we report the “normalized performance” representing the average of the z-score across the 22 tasks. The z-score is computed based on the model’s performance relative to the leaderboard for a task, adjusted for the polarity of the task metric, i.e., multiplied by -1 if “lower is better”.
Molecules vs. language
In the context of LLMs, our scaling experiment results may seem surprising given our models are only trained on a few million data points (molecules) while LLMs are typically trained on larger datasets comprising trillions of tokens. To better understand the performance increase and gap in dataset size, it’s helpful to draw a few analogies between molecules and language.
In our setting, molecules are analogous to sentences in language processing, while the atoms and bonds are analogous to tokens. Moreover, the task is supervised and some molecules have thousands of associated labels coming from experimental data. This allows the learned molecular embeddings to be much richer than such derived from simply retrieving a missing token.
Conclusion
In conclusion, our work presents the first work in scaling molecular GNNs to the billion parameter regime, with consistent performance gains on downstream tasks with increasing model scale. This is consistent with recent advances in natural language processing and computer vision, and effectively brings us to the era of foundational models for molecular representation learning.