


Paper link: https://arxiv.org/abs/2310.03223
Code link: https://github.com/tsa87/TacoGFN-SBDD
_________________________________________________________________________
Recently, we have seen tremendous success with diffusion models for image and video generation, with the advent of models such as Stable Diffusion, and Sora. Diffusion models have also been successfully applied to protein generation, and unconditional molecule generation. Given sufficient data, diffusion models can generate realistic and new data points, similar to training data under the maximum likelihood estimation (MLE) objective.
Models like Stable Diffusion and Sora have billions of diverse images/videos to train on. In structure-based drug discovery, where we want to design small molecules with desirable properties and binding to a pocket in a target protein, we have many orders of magnitude less data. For example, the PDBBind dataset, the most comprehensive data collection attempt, only has ~19,000 protein-ligand complexes!
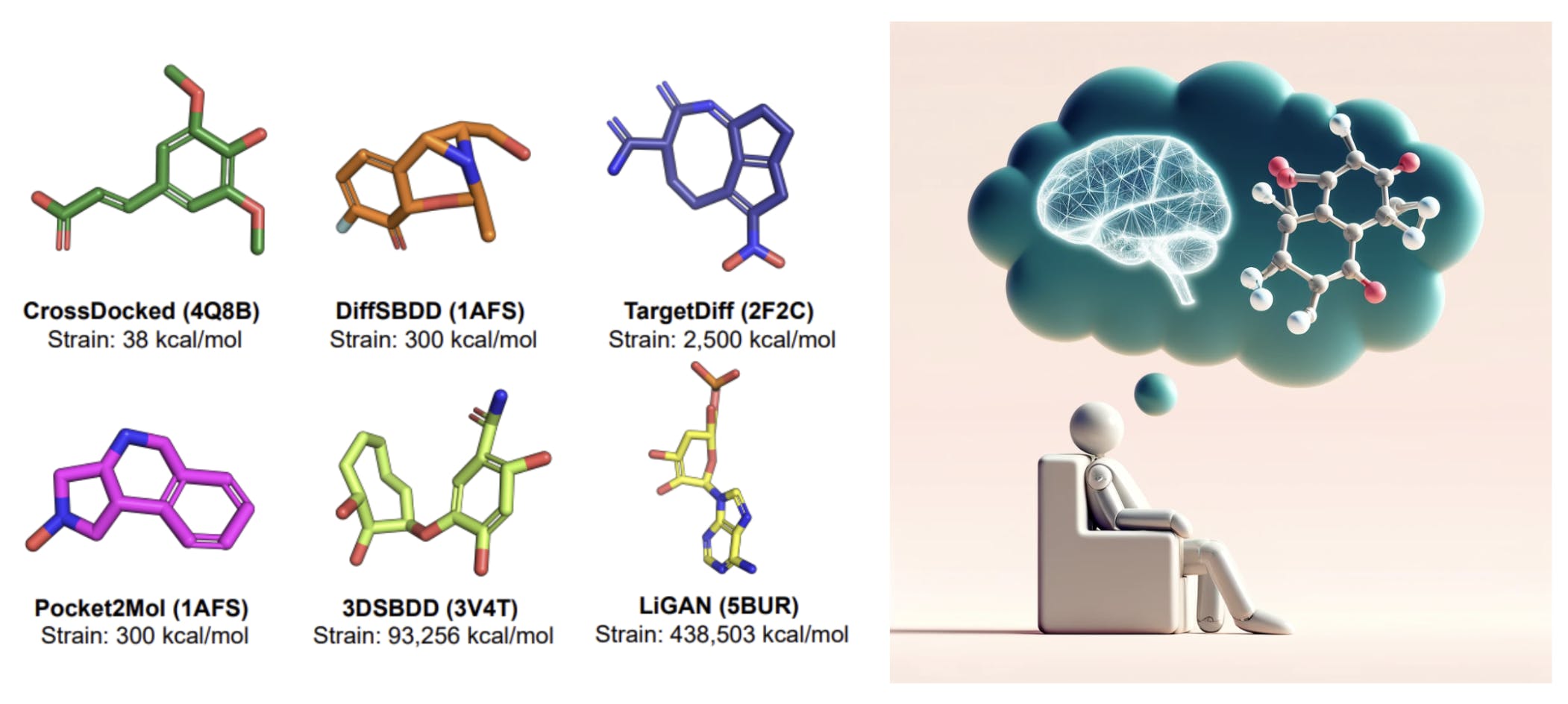
A recent line of work has recruited diffusion models with MLE objectives for structure based drug design. However, this limitation of dataset size is a real bottleneck for MLE based generative models to be successful for real-world drug design settings (more on this in a sec!).
Should we then simply give up because there’s not enough data? Of course not! We have a tremendous opportunity to discover new drugs at scale with the help of machine learning and it’s too important of a problem to give up. Plus, we believe the limitation of dataset size in structure-based drug design can be overcome with an alternative approach!
Currently, everybody uses this MLE training objective on this tiny dataset, but we feel like that objective doesn't align well with the goals of structure-based drug design. We'll tell you why, and suggest an alternative approach that could be the key to unlocking the structure-based drug design problem.
What's wrong with an MLE objective?
Diffusion models are trained to maximize the likelihood (MLE) of observing the training data, so we expect them to generate similar molecules to the training set. This is great if you have a large and diverse training set, but can be a problem if you don't.
Unfortunately for structure-based drug design, the dataset contains only a tiny fraction of the estimated drug-like chemical space of 10^60 molecules! Many molecules in the PDBBind dataset aren't easily synthesizable or don't have the necessary drug-like properties. In fact, when you remove common biomolecules (such as Sulphate - SO4, Nucleosides - NTP/NDP/NMP) and duplicates from PDBBind you are only left with ~4,200 unique drug-like molecules as examples. With models trained with MLE, we will mostly likely be stuck with molecules with unsuitable properties and high-resemblance to training set molecules.
Instead, how can we freely explore the entire chemical space, and find molecules that satisfy our affinity and property goals? We can use GFlowNets to learn a policy for constructing molecules!
GFlowNets are a recently proposed class of generative models trained to sample objects proportional to a reward function. GFlowNets overcome this dataset size limitation of MLE by generating new examples and learning from their given reward. As a result, the quality of training only depends on the compute budget of calling the reward function (and the accuracy of the reward function). This RL formulation also allows us to specify our preference for molecules based on their affinity, drug-likeness, and synthesizability properties. With GFlowNets we can explore a much greater chemical space and find high-reward modes. Empirically, our proposed approach improves the current hit rate for structure-based drug design from 7.94% to 52.63%.
Target Conditioned GFlowNet for structure based drug discovery (TacoGFN)
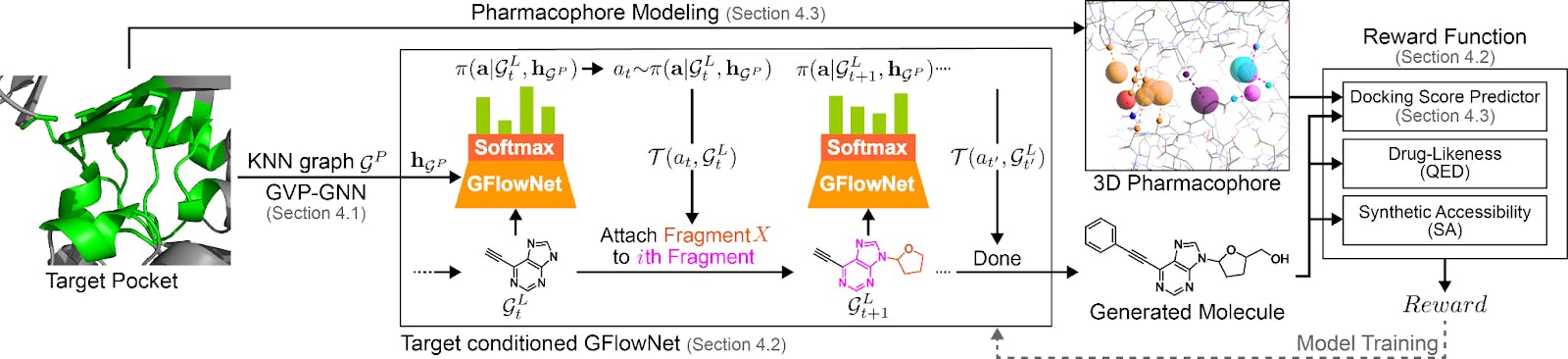
In our recent preprint, Target Conditioned GFlowNets (TacoGFN) address the problem of limited training dataset by instead generating molecules. We modify GFlowNet to generate molecules conditioning to any arbitrary protein pocket structure. At a high level TacoGFN contains three modules:
Pocket encoding module represents the 3D pocket structure as the pocket conditioning. This is implemented by GVP-GNN (geometric vector perceptrons), which preserves the geometric information of the pocket structure.
Molecular generation policy module learns a molecular construction policy conditioned on pocket information and generates molecules with probabilities proportional to their reward (instead of data distribution as in MLE).
Multi-objective reward module takes the generated molecule and computes a reward that consists of the molecules’s drug-likeness, synthetic accessibility and the predicted binding affinity with respect to the target protein pocket. Since we don’t have an infinite compute budget to run molecular docking on every molecule generated, we train a proxy machine learning model to predict the docking score.
At a high level, TacoGFN learns from continuous “trial and errors”, where the molecular generation policy module continuously tries to generate new molecules with more desirable properties by learning from the feedback of the reward module. This is different to the MLE paradigm, where the model tries to “memorize” the few existing examples.
RL shows promising results for structure-based drug design
We looked at how well TacoGFN does in comparison to MLE-based generative models on the popular CrossDocked-2020 benchmark. TacoGFN significantly outperforms existing generative models with the MLE objective in terms of novelty, docking score, drug-likeness, synthetic accessibility and overall hit rate.
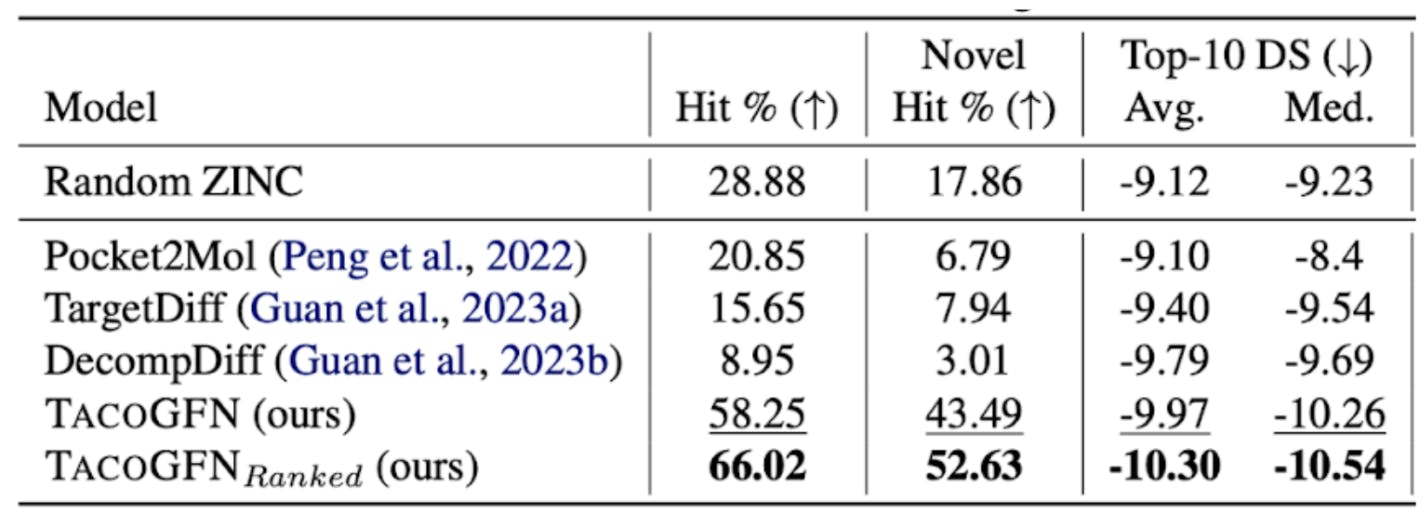
Is MLE all you need for SBDD?
In this blog, we showed that using MLE as the sole training objective is insufficient and misaligned with the goals of SBDD. To address the limitations of MLE, we have framed structure-based drug discovery as a multi-objective RL task. TacoGFN explores the chemical space to generate novel molecules with high binding affinity and desired properties and shows very promising results compared to previous MLE-based methods.
While MLE may not be all you need for SBDD, we don’t claim RL is all you need for SBDD either. In fact, we think that many generative models will actually benefit from the combination of MLE and RL. These models will not only learn to model the data distribution but also shift the generated distribution towards areas with high reward. By retaining the MLE objective, we ground the model on real-life examples. By allowing generative models to interact with an environment which provides rewards based on the quality of the generation, we can better align these models to produce useful and controllable outputs for real-world problems.
After all, we may just need multiple keys to unlock the problem of structure-base drug design.
Paper, code and contact
For many more details and results, see our preprint: Shen T, Seo S, Lee G, Pandey M, Smith JR, Cherkasov A, Kim WY, Ester M. TacoGFN: Target-conditioned GFlowNet for Structure-based Drug Design. (2023) and checkout our official github repo.
Please feel free to reach out to us by email if you have any questions! You can also contact us on Linkedin:
Seonghwan Seo: https://www.linkedin.com/in/seonghwan-seo-5199b2251/
_________________________________________________________________________
*: PoseCheck