

Drug discovery is really difficult. Finding a molecule that treats a disease is already a hard problem, but a successful drug can’t just treat the disease. The molecule also has to be absorbed by the body, find its way to the diseased area, exert its effect, and then exit the body all without causing any major damage along the way. Measuring all of these necessary druglike properties in the lab is really slow and expensive, so it would be fantastic if we could predict them computationally.
We built ADMET-AI to solve this problem. ADMET-AI is a machine learning platform that predicts the absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties of a molecule, which are many of the properties needed in a safe and effective drug. We wanted to make ADMET-AI as accessible as possible to the research community, so we made a free website, wrote a paper, and made the code open source as a pip-installable Python package. Please give it a try, and read on for more details!
Developing ADMET-AI
In the past, we’ve had success using graph neural networks (GNNs) to predict all sorts of properties of molecules, from quantum mechanical energies to human toxicity. So for ADMET-AI, we decided to use the same model architecture, called Chemprop-RDKit, to predict ADMET properties.
How Chemprop-RDKit works
Chemprop-RDKit (Figure 1) contains a GNN called Chemprop that computes simple features for each atom (e.g., atom type) and each bond (e.g., bond type) and then runs several steps of message passing with neural network layers to aggregate atom and bond information across the molecule to build a single representation of the whole molecule. This representation is then concatenated with 200 molecular features computed by RDKit, and the combined representation is passed through several feed-forward neural network layers to predict molecular properties. With this hybrid architecture, Chemprop-RDKit combines the strength of GNNs (understanding local neighborhoods of the molecule) with the power of RDKit (computing features that describe the entire molecule).
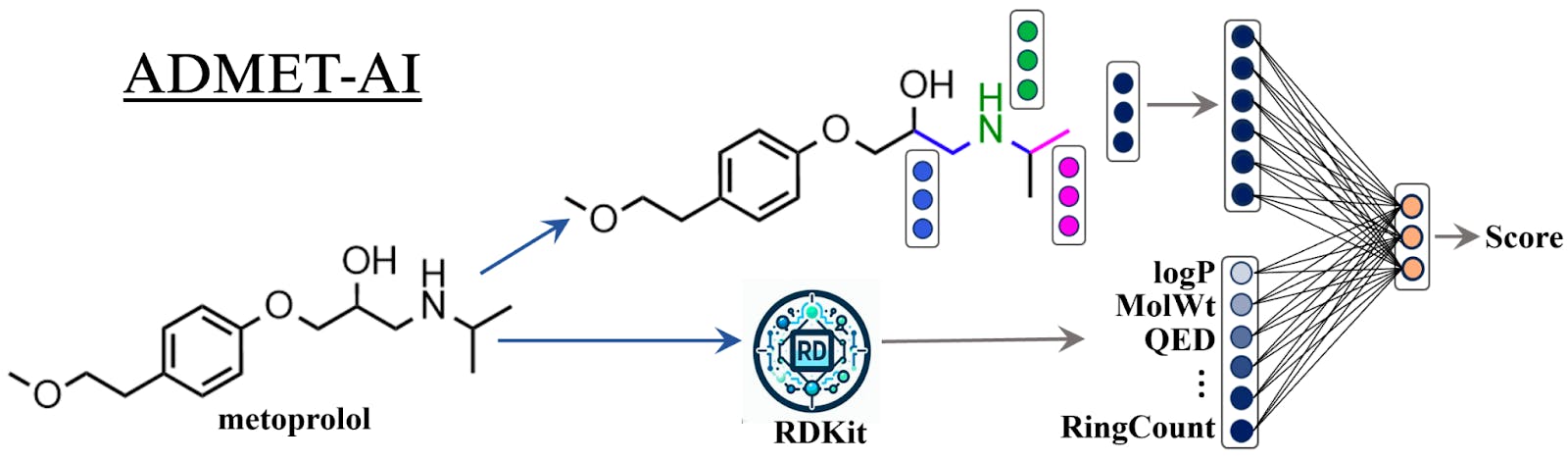
Training on benchmark ADMET data
We trained ADMET-AI’s Chemprop-RDKit models using ADMET data from the Therapeutics Data Commons (TDC), which is a great resource of publicly available datasets for drug discovery. The TDC has a leaderboard called the ADMET Benchmark Group with 22 ADMET datasets, and this leaderboard is perfect for comparing different machine learning models for ADMET prediction.
Since we wanted to build the most comprehensive ADMET prediction model possible, we also used an additional 19 ADMET datasets that are in the TDC but are not included in the leaderboard for a total of 41 ADMET datasets (10 regression, 31 classification). We also built two multi-task ADMET datasets, one with all 10 regression datasets and one with all 31 classification datasets, so that we could train multi-task models since they’re often faster and more accurate than training many single-task models.
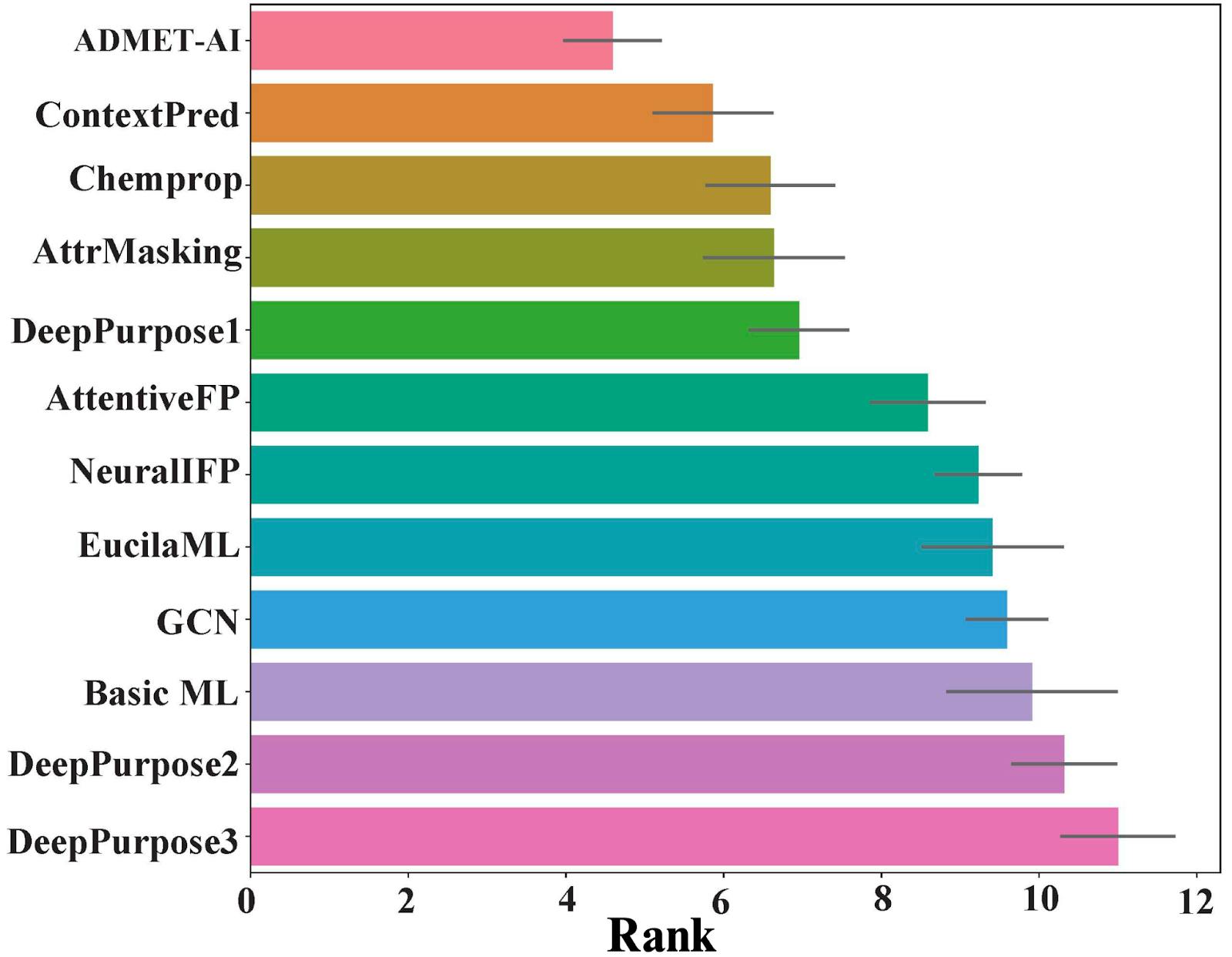
ADMET-AI outperforms competitors
We first tested ADMET-AI on the 22 datasets from the TDC ADMET leaderboard. We were excited to see that ADMET-AI had the best average rank compared to all other models (Figure 2). While other models are often accurate on only a few ADMET properties, ADMET-AI is the best on average across all the ADMET properties, so it’s ideal for predicting the entire spectrum of ADMET properties at once.
We then tested ADMET-AI on the full set of 41 TDC ADMET datasets. We saw that ADMET-AI worked well both as 41 single task models (one per dataset) and as two multi-task models (one for all regression datasets, one for all classification datasets). Since running two multi-task models is much faster than running 41 single-task models, we decided to use the multi-task models in our website and Python package.
Building the ADMET-AI Website
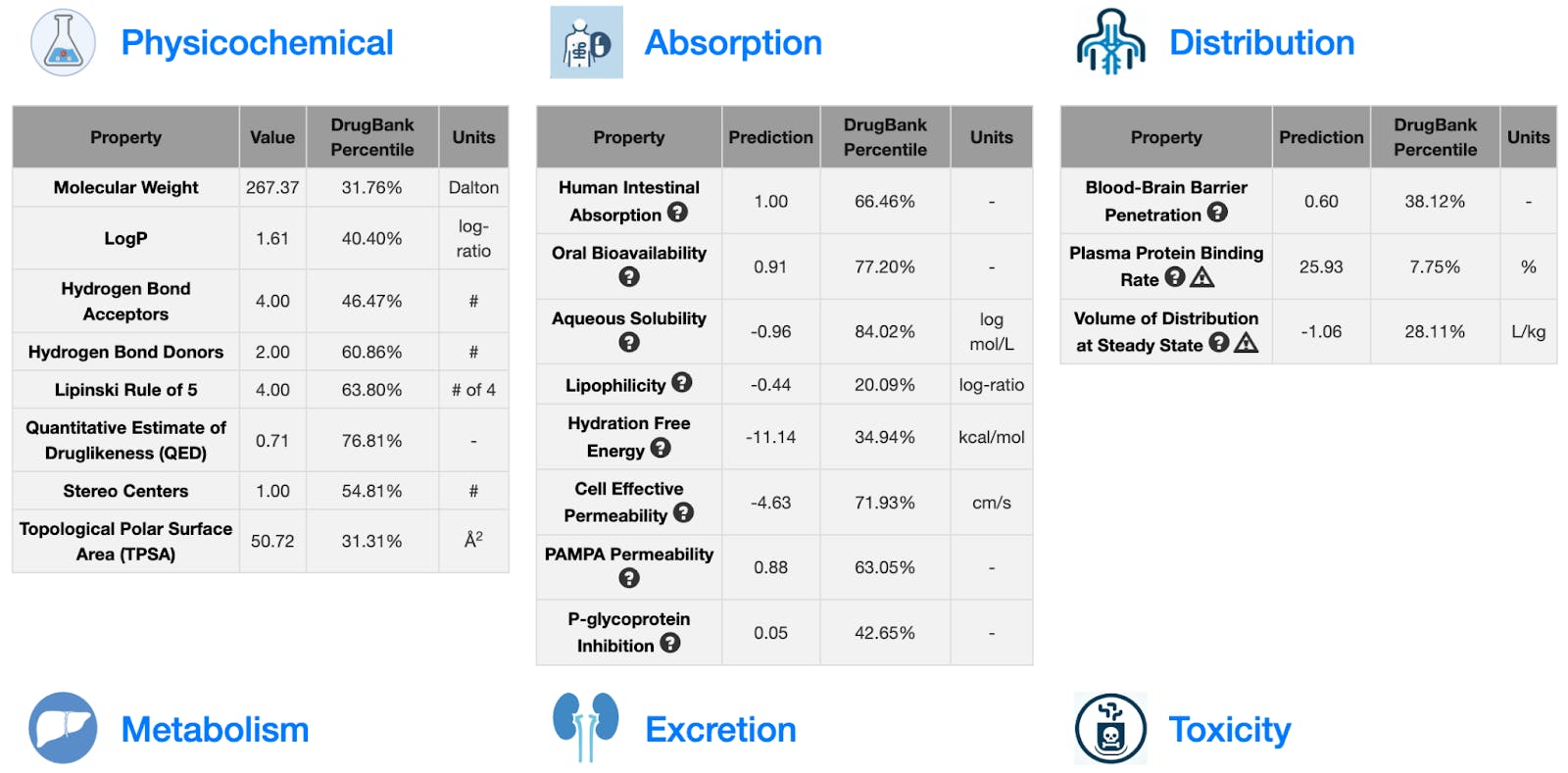
We wanted to make fast and accurate ADMET prediction easily accessible to researchers regardless of computational background, so we built the user-friendly ADMET-AI website. If you visit the website, you can input up to 1,000 molecules at a time by entering SMILES, uploading a CSV file, or drawing molecules with an interactive tool (Figure 3). You can then make predictions and view them on the website or download them as a CSV. The website displays both a radar plot summary of five key druglike properties (Figure 4) and prediction values for all 41 ADMET properties (Figure 5).
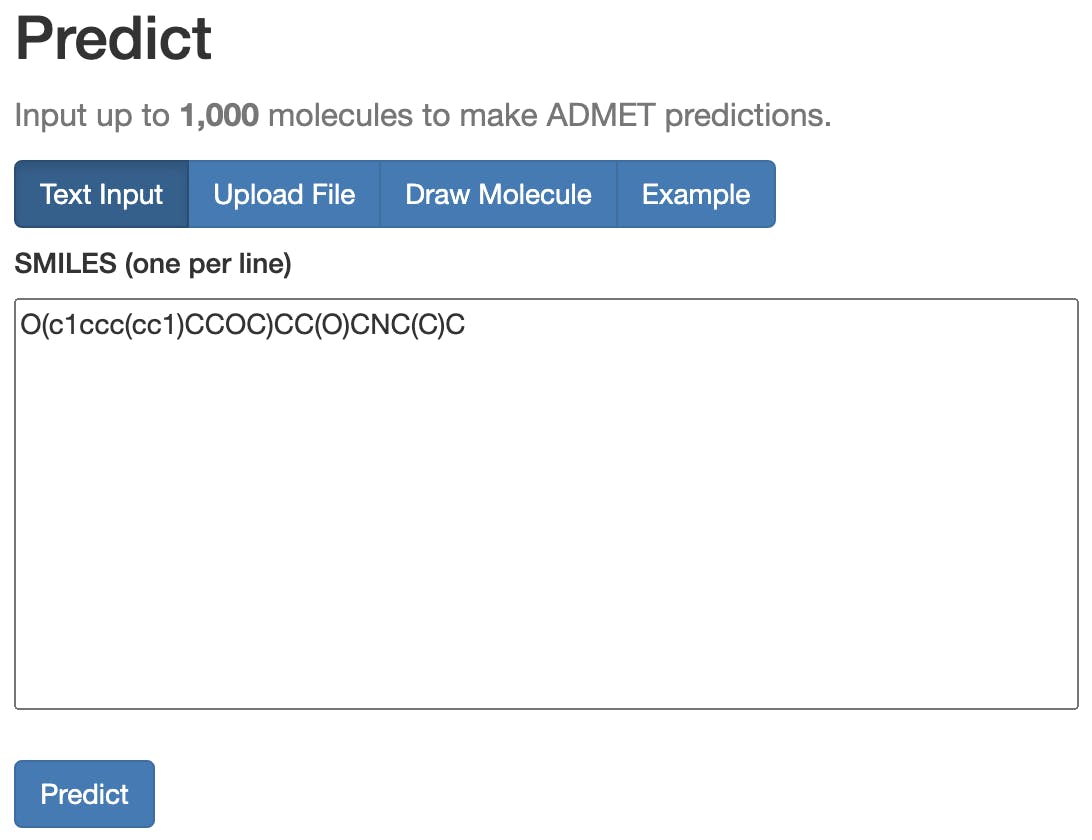
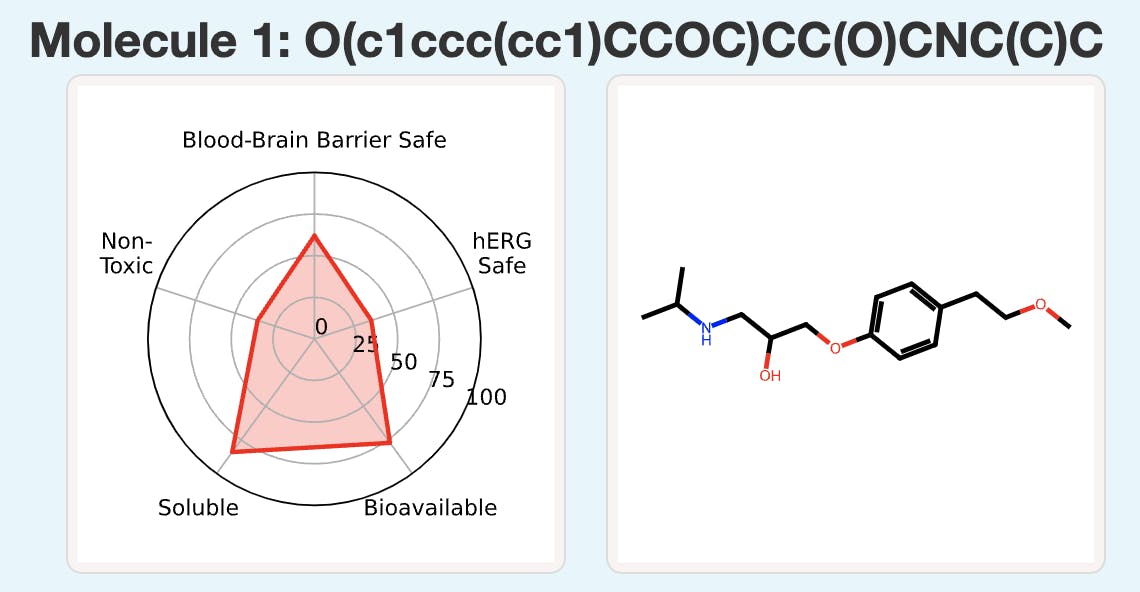
There are several other publicly available ADMET prediction websites, such as SwissADME and ADMETLab 2.0, but ADMET-AI stands out with three key differences: (1) contextual predictions, (2) local predictions, and (3) speed.
Contextualizing ADMET Predictions
ADMET predictions can be difficult to interpret in isolation because different categories of drugs have different requirements on their molecular properties. For example, high toxicity is acceptable as a side effect of chemotherapies since there are often no better alternatives, but high toxicity is generally unacceptable for drugs like antibiotics since toxicity can outweigh the benefits of the drug, especially compared to safe alternatives.
We thought that we could provide context for ADMET-AI’s predictions by comparing predictions on new molecules to predictions on known drugs. To do this, we created a reference set of 2,579 drugs from the DrugBank that have obtained regulatory approval. We then applied ADMET-AI to make ADMET predictions on all of these molecules. Now, when ADMET-AI makes predictions for a new molecule, it also computes the percentile of the ADMET predictions with respect to the 2,579 reference approved drugs (Figure 5). For example, if a molecule receives a toxicity percentile of 90%, it means that the molecule is more toxic than 90% of approved drugs.
We wanted to provide users with even more fine-grained context, so we also added an option to let users pick an Anatomical Therapeutic Chemical (ATC) code, such as “antibiotics for topical use,” to create a more refined reference set for computing percentiles (Figure 6). If you select an ATC code, you’ll then see your proposed molecule compared to only approved drugs labeled with that ATC code, so you can see if your molecule shares the same range of ADMET properties as approved drugs for the same disease.
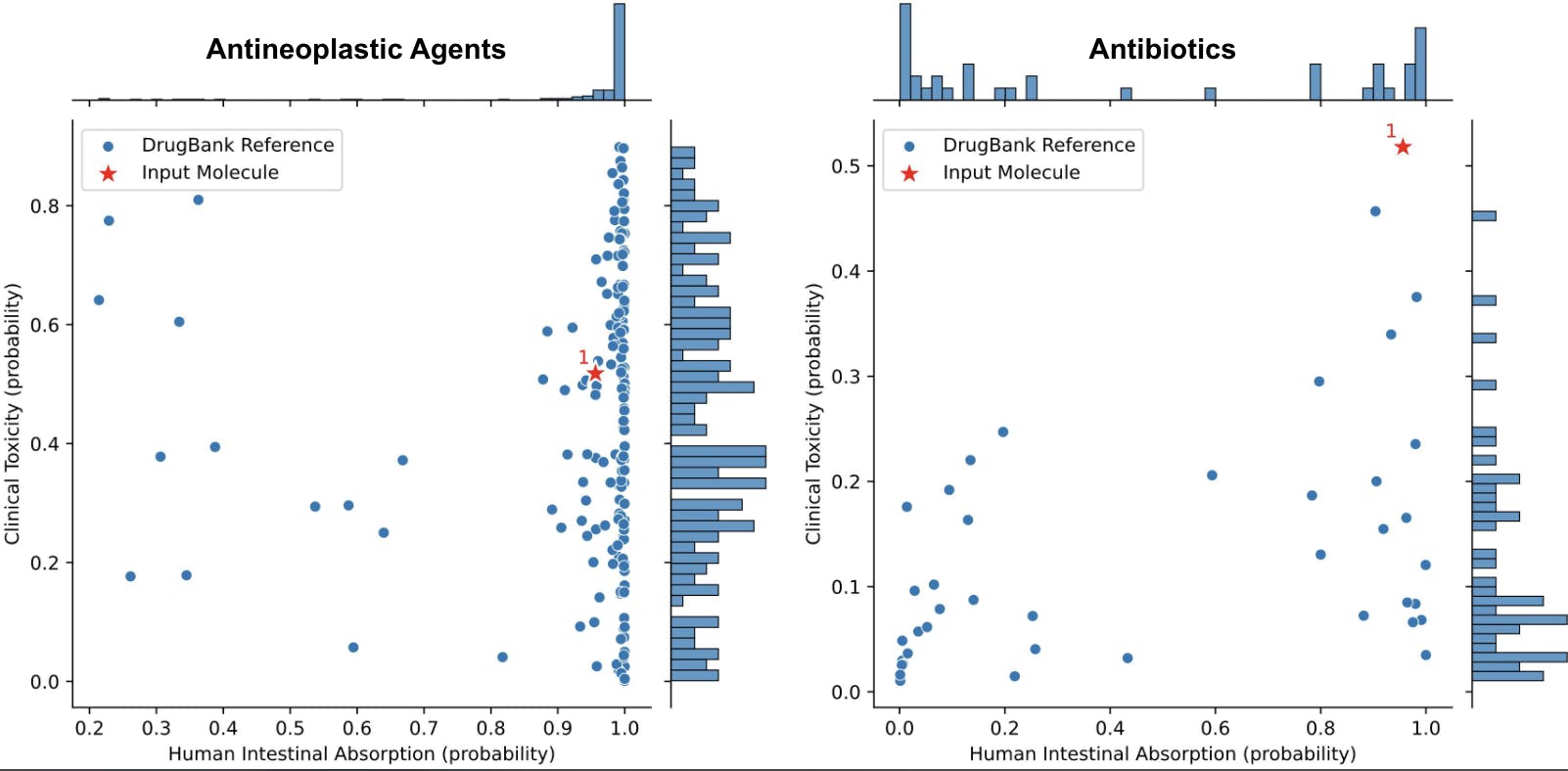
To make it easier to visualize this information, ADMET-AI also creates a scatterplot that compares the input molecules to the reference DrugBank molecules based on two user-selected ADMET properties (Figure 7).
Local Predictions
We made the ADMET-AI website easy to use, but for more computationally oriented users, ADMET-AI is also available as a Python package for making predictions locally. The ADMET-AI package contains the same models as the website but can be run on faster hardware (e.g., a GPU and more CPUs) with no limit on the number of molecules per prediction. You can install ADMET-AI with pip install admet-ai, and then you can make ADMET predictions using the admet_predict command-line tool or by importing the ADMETModel class in Python.
# Installation
pip install admet-ai
# Command line interface
admet_predict --data_path /path/to/smiles.csv# Python module
from admet_ai import ADMETModel
# Load ADMET-AI model
model = ADMETModel()
# Predict on one molecule => dictionary
admet_predictions = model.predict(smiles="CCO")
"""
{"hERG": 0.01, ..., "ClinTox": 4.79e-04}
"""
# Predict on multiple molecules => Pandas DataFrame
admet_predictions = model.predict(smiles=["CCO", "CC"])
"""
hERG ... ClinTox
CCO 0.01 ... 4.79e-04
CC 0.11 ... 5.40e-06
"""Speed
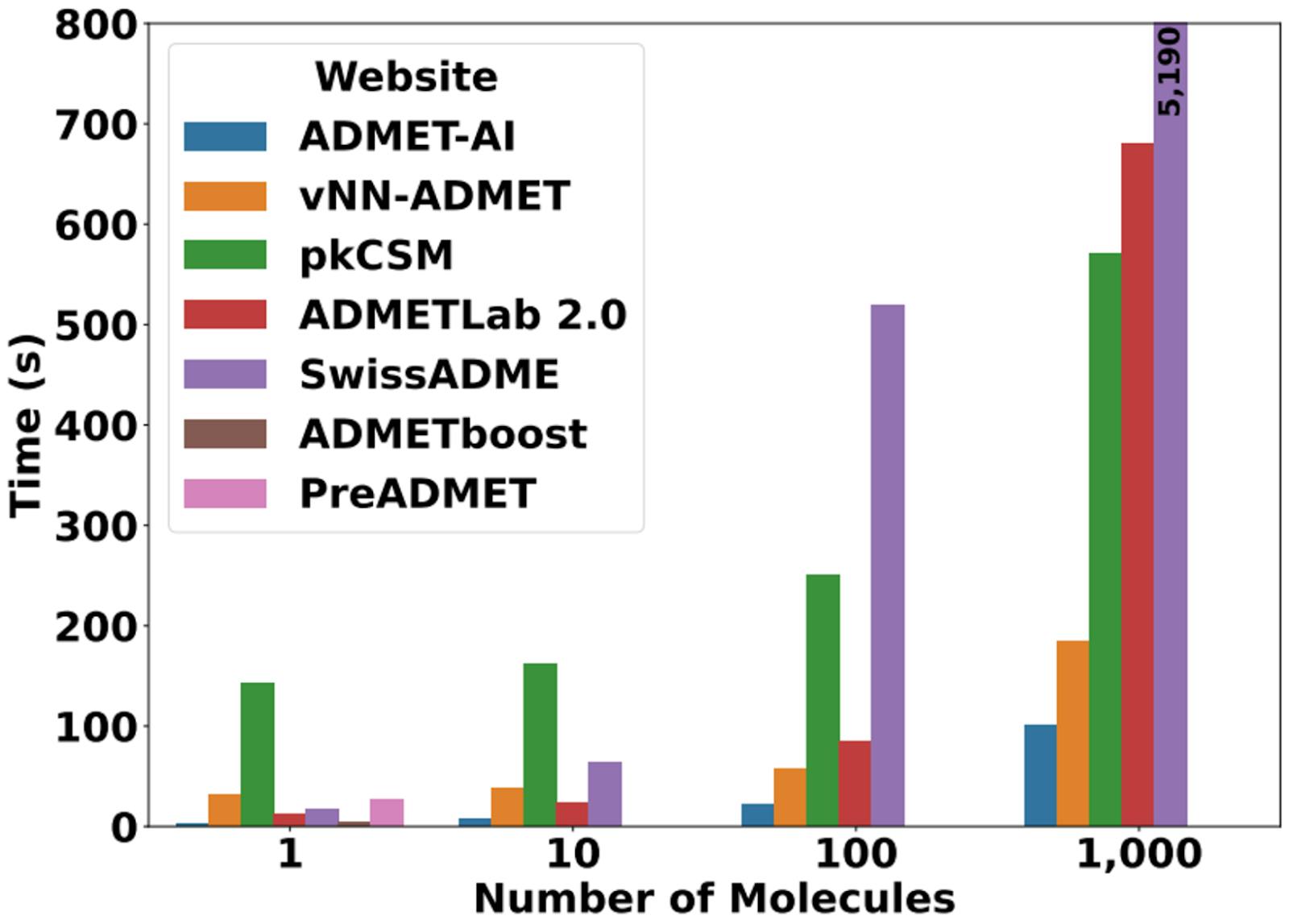
One big benefit of ADMET-AI is that it’s much faster than other publicly available ADMET prediction tools. ADMET-AI can make predictions on 1,000 molecules in just 1 minute and 41 seconds compared to over 3 minutes for the next fastest model (vNN-ADMET) and 1 hour and 26 minutes for SwissADME (Figure 9).
Users can also run ADMET-AI locally with more powerful hardware for even greater speed. On a local machine with 32 CPU cores and a GPU, ADMET-AI can make predictions on one million molecules in just 3.1 hours. This means it’s realistic to make ADMET predictions on large-scale chemical libraries for drug screening using ADMET-AI.
Conclusion
ADMET-AI is a simple, fast, and accurate platform for ADMET prediction with both an easy-to-use website and a powerful Python package. ADMET-AI is the most accurate model on average across the TDC ADMET leaderboard and is significantly faster than all other publicly available ADMET websites. It also uniquely provides context for ADMET predictions by comparing the ADMET properties of new molecules to those of approved drugs in relevant therapeutic categories. As a fully free and open-source platform, ADMET-AI can be a powerful drug discovery tool for identifying compounds with favorable ADMET properties for further development. We hope you’ll try ADMET-AI, and please feel free to reach out with any questions or feedback!