

Proteins are complex machines, fundamentally no different than a car or a printer but much smaller. Their function is inherently tied to their 3D shapes, and if we can understand and generate protein structures we could advance drug discovery and synthetic biology. Recently, diffusion models have emerged as a powerful generative model across domains including images, videos, small molecules, and proteins. It's still challenging to generate high-quality proteins efficiently, but in this blog post we'll tell you about the progress we've made leveraging Latent Diffusion Models (LDMs) for this purpose. The overview diagram is shown below:

Our method reduces the complexity of protein modeling by training an SE(3) equivariant protein autoencoder to encode protein structures from input space to latent space. Then we use the diffusion model to capture the distribution of natural protein structures in the condensed and information-rich latent space. If you just used the diffusion model (which operates in the protein space), generating 1000 protein structures would take about 4 hours, but it only takes 15 minutes to generate them in the latent space and then map to the protein space.
How to Design a Protein Autoencoder
In computer vision, variational autoencoders (VAEs) have been highly effective. With VAEs, you can effectively compress images to smaller sizes (as a latent representation) without losing the critical essence depicted in the picture. For instance, an image of 128x128 pixels can be compressed to a significantly smaller latent representation, such as 64x64 or even 2x2 pixels, while still keeping track of the details that connect different parts of the image together. This feat is achievable due to the grid-like structure of images, where each pixel is intricately linked to its adjacent pixels.

However, when we move from the 2D realm of images to the 3D domain of objects like proteins or atoms, things can get more complex. Unlike images that display pixels in a consistent and predictable arrangement, 3D objects resemble intricate puzzles, with pieces (atoms or amino acids) that can be connected in many different ways to form chemical bonds. Downsizing these 3D structures by decreasing the number of components risks omitting critical information about the interconnections. Simply put, excessively reducing the components might render the original structure irrecoverable.
But there's a silver lining when it comes to proteins, particularly their alpha carbon level backbones. Proteins have a unique feature: their backbones can be thought of as sequences, similar to strings of text, where each "letter" (amino acid) connects only to its immediate neighbors. This sequential attribute gives us a clue about how to simplify protein structures without losing the essential connections between amino acids.
Leveraging this insight, our 3D equivariant protein autoencoder considers both the amino acid sequence and 3D geometric information of protein backbones. For example, within each downsampling layer, we construct a graph that contains the input nodes (white) and initialized downsampling (black), and then perform equivariant message passing on the constructed graph. The edge connections between input nodes and downsampling node are similar to how we define the receptive field or kernel size in a 1D sequence. After the message passing, only updated downsampling nodes will be kept as input in the next layer.
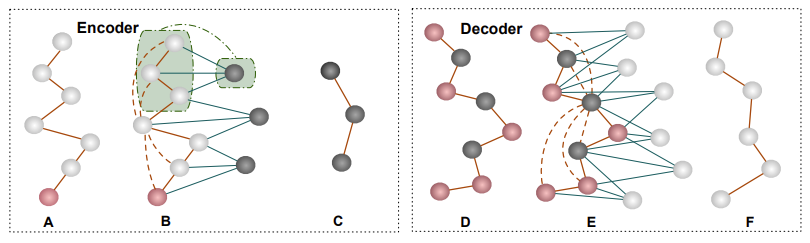
This technique allows us to effectively reduce the complexity of protein structures while preserving the vital connectivity information between amino acids. By considering the protein backbone as a sequence, akin to a sentence where each word (amino acid) connects to the next, we can simplify the structure without losing the connection information.
Learning a Diffusion Model in Latent Space
Reducing the complexity of protein backbones helps improve the effectiveness and efficiency of diffusion models. Here's why focusing on a smaller and more manageable modeling space can be advantageous:
1. Learning Effectiveness: Just like it's easier to work with a smaller, simpler puzzle than a larger, more complex one, diffusion models learn more effectively when dealing with simpler data. For example, creating digital images of a smaller size (like 128x128 pixels) is much simpler than dealing with larger images (such as 1024x1024 pixels). Similarly, by simplifying the structure of protein backbones, we provide the diffusion model with a more manageable and structured latent search space, enabling it to learn the essential features and patterns of protein structures more efficiently.
2. Generation Efficiency: Generation in latent space can improve memory efficiency as the latent space is much smaller than the protein space. For the same amount of GPU memory, more proteins can be sampled in latent space than in protein space. In drug discovery, we often need to screen lots of candidates to find “good” ones. For a generative model, we want to be able to generate as many samples as possible. In this sense, parallel sampling in latent space could significantly improve efficiency. You can find more experiments on parallel sampling efficiency in our paper.
Once we've simplified the protein backbones into a more compact form using our pre-trained protein autoencoder, our goal is to build this condensed information into a simple prior distribution that we can easily sample from, such as Gaussian distribution. This step involves using advanced diffusion models specifically tailored for latent protein structures. We adopt a similar architecture with EDM, a diffusion model on small molecule generation. A key part of this process is ensuring the distribution SE(3) invariance, which means that the probability of sampling a protein should be independent of rotation and translation transformation. This is achieved by moving latent protein structures to zero centroids and employing a rotation equivariant reverse diffusion process. Proteins only contain right-handed alpha helices, so the denoising network and protein autoencoder should not be equivariant to reflection.

Structure and Sequence Co-Design
Another key difference from other protein generation methods is that our method can perform structure and sequence co-design, as the decoder of the protein autoencoder can predict amino acid types. Specifically, we can use decoded sequences as the generated protein sequences instead of predicting sequences from decoded structures using inverse folding methods. The most common pipeline in protein generation methods is generating structures first by the model, and then using another pre-trained inverse folding model such as ProteinMPNN to predict amino acid sequence from generated structures.

Self-consistency is important in protein generation because we want the designed amino acid sequence to be able to fold into the generated protein structure. We found that amino acid sequences predicted from generated protein structures using the pre-trained inverse folding model are more consistent with generated structures than those decoded from the protein autoencoder. This is because conditionally predicting sequences is easier than jointly generating both structures and sequences together. However, even using generated sequences, LatentDiff can achieve similar results with earlier protein generation methods such as ProtDiff.
Future Directions
Where can we go from here? There are several potential directions for future work:
(1) Adjust the 3D protein autoencoder to support arbitrary length input and generate arbitrary length protein backbone structure.
(2) Train the 3D protein autoencoder on more protein structures. We were limited by computing resources, so we couldn’t train our 3D protein autoencoder on all protein structures predicted by AlphaFold (AlphaFold DB).
(3) Improve the structure and sequence co-design; this will be easier with the more powerful protein autoencoder we get from steps (1) and (2). A powerful and robust autoencoder is very important in latent diffusion, as can be seen from stable diffusion.
(4) Extend our method to full-atom backbone structure generation and increase the length of generated proteins.
Conclusion
In our work, we propose a latent diffusion model to generate protein representations in the compact latent space and then decode them into the protein space via a pre-trained protein autoencoder. LatentDiff is effective and efficient in generating designable protein backbone structures and we hope this can inspire more powerful latent diffusion methods in the area of protein generation.
If you have any questions, check out our paper and feel free to reach out to us!